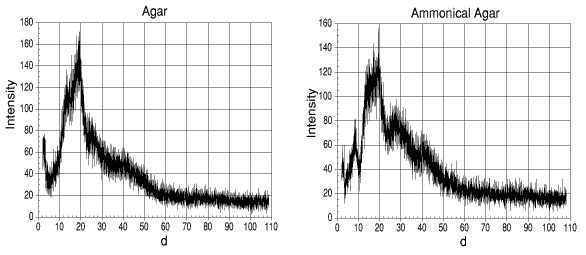

Fig. 1 XRD Scans.
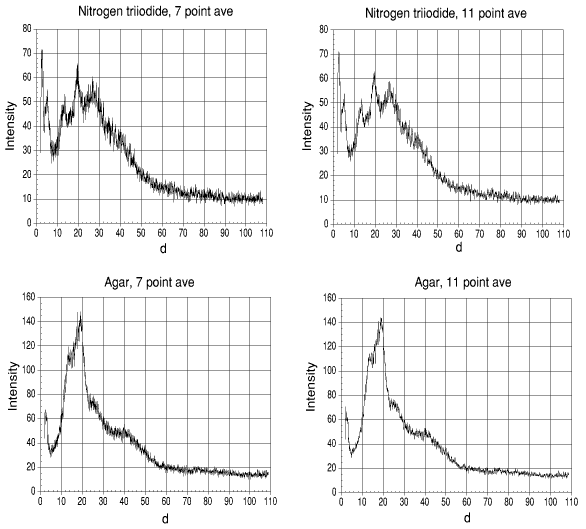
Fig. 2, the moving average method of "cleaning" data.
1.6 X-ray diffraction results.
The X-ray beam has a power rating of 1600W (40kV, 40 mA). The samples were scanned between d=2° and d=108° at 0.2° intervals. The sample holders were also scanned as they may have interfered with the overall scans. The figures resulting from the sample holders could then be subtracted from the result.
It can be seen from the scans (fig. 1) that there was a high level of noise on the scans. A statistical method of "cleaning" the results is that of a moving average.
The moving average method was applied by taking the average of seven intensity values (three above the selected figure, the selected figure and the three after the selected figure - the selected figure was any figure selected which had a minimum of three numbers before it at the start, e.g. point 4 would be the start selected figure, but point 3 could not be the start).
This moving average was performed for all the intensities of the range of d values. The result can be seen in fig. 2 (as well as what was produced when the moving average was increased to 11).
It can be seen that while the signal is "cleaned" very well, there is an obvious loss of accuracy. A moving average will invariable mean that some points will be removed. The higher the moving average number, the less the accuracy.
Fig. 1 XRD Scans.
Fig. 2, the moving average method of "cleaning" data.
Even with the "cleaning" of the data, it was still not possible to obtain points which could be fed into a molecular modelling package, or converted into an ORTEP file for modelling.
On close examination of the gels, it was clear that there was a problem with the method used.
Each of the gels had been disrupted from being smooth to becoming a ridged powder and there had been enough power to cause a series of small detonations in the stabilised sample or possibly the scan time had been too long, causing the gel to be shaken by the vibrations. from the XRD.
The X-ray exposure time could have been enough to have disrupted the gel at a molecular level and cause the agar to break up.
Though the data was not of sufficient quality to be able to determine an absolute structure, there were a number of similarities between the scans to be able to try to assign regions to common constituents.
The holders have been excluded as it was unlikely they had contributed to the result.
With the exception of the NI3 agar, all of the agars had a number of similar points at around d = 20°, with a small hump around d = 30°.
With the NI3 agar, there is still a number of points around d = 20°, but there is a significant hump prior to this at around d = 10°. As this had not appeared in any of the previous scans, it may be possible to conclude that this had been due to the NI3 within the agar matrix.
There were a number of other assignments which could be made. There are a number of similar peaks for the all the samples of agar other than those previously noted.
0.880 S.G. ammonia contains 31% w/v ammonia, the rest being water. The humps at between d = 27° and d = 45° can be attributed to water as they appear in all of the samples with the exception of the agar powder. Peaks which can be attributed to the ammonia are at around d = 8°. These are observable in both the NI3 agar gel and the ammonical agar gel (though to a lesser extent in the NI3 agar).
From the evidence of the other peaks, it can be assumed that the high intensity peaks at around d = 2° and d = 5° as well as the broad range of peaks between d = 15° and d = 30° can be attributed to NI3 with a contribution in this area from agar. The degree of contribution could not be resolved as the interference caused in the signal for the NI3 and that of the agar is not known.
